Low aqueous solubility is an increasingly common and major challenge in drug development. Advances in in-silico drug design and the use of high throughput screening have led to more potent and targeted molecules; however, these tend to be hydrophobic, creating a significant hurdle for getting these drugs into the body. Low aqueous solubility will lead to low bioavailability and thus performance of pharmaceuticals. This is particularly important for oral formulations, which is the preferred administration route for pharmaceuticals due to high patient compliance and ease of administration. The Biopharmaceutical Classification System (BCS) is used as a tool to differentiate drug candidates based on their solubility and permeability. For between 70 and 90 % of drugs in development, it is estimated that bioavailability is limited by low solubility.
Overcoming The Solubility Challenge Using Crystal and Particle Engineering – Piroxicam, A Case Study.

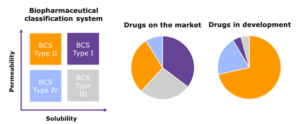
Figure 1: The Biopharmaceutical Classification System differentiates oral drug candidates based on their aqueous solubility and permeability relative to clinical dose: BCS Class I (high solubility, high permeability), Class II (low solubility, high permeability), Class III (high solubility, low permeability) and Class IV (low solubility, low permeability). The pie charts show the estimated break down of drugs on the market and drugs in development into each of these classes. It is estimated that as many as 90% of all drugs in development suffer from low bioavailability as a result of low aqueous solubility.1
There are a growing number of proven approaches to tackle this solubility challenge. Unfortunately, there is not one method that is the best fit for every drug. The most suitable approach will depend on the compatibility with the active pharmaceutical ingredient (API), target dose and administration route. It is well recognized that the earlier this issue is tackled, the better. Veranova has investigated and compared a number of different crystalline form and particle engineering techniques to improve the bioavailability of a non-steroidal anti-inflammatory drug, piroxicam.
When developing a new compound, the first step is rigorous physiochemical profiling to guide the process. Piroxicam falls into BCS Class II (bioavailability limited by aqueous solubility). Although piroxicam is well characterized in the literature, we collected our own in-house profiling data. Piroxicam was confirmed to have a basic pKa of 5.3 and an acidic pKa of 1.9. The LogP of piroxicam was measured to be 1.8. The benefit of collecting data in-house is not having to rely on historical literature values which can sometimes vary from the true value. We collected thermodynamic solubility (24 hours) data for piroxicam in phosphate buffered saline at pH 7.4, above this basic pKa. The thermodynamic solubility was shown to be 0.4 mg/ ml.
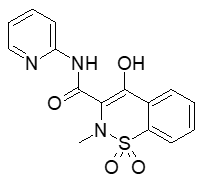
Figure 2: Structure of piroxicam, a marketed non-steroidal anti-inflammatory agent which is BCS Class II.
Salt formation is an extremely widespread method for addressing low aqueous solubility. An ionized form is expected to have higher aqueous solubility than a non-ionized form. This would generally be the first port of call to investigate provided that the molecule has generally accessible acidic or basic functional groups. Roughly half of all FDA approvals are of drugs in their salt form.2 Our data shows that piroxicam has an acidic group with an accessible pKa. A known ethanolamine salt of piroxicam was generated by Veranova.
Cocrystal formation is increasingly used as an approach to improve physiochemical properties. Although the absolute thermodynamic solubility of drug molecules cannot be changed by the preparation of cocrystals, certain API cocrystals display an improved kinetic solubility behaviour and Intrinsic Dissolution Rate (IDR) that can lead to improved bioavailability. A known cocrystal of piroxicam with succinic acid was prepared.
As well as manipulating the solid form, particle engineering can be used to enhance the performance of a poorly soluble drug. Particle size reduction is one of the simplest methods for enhancing the free powder dissolution rate. It works by increasing the surface area available for dissolution. Our FPS spiral jet mill (Labomill-1) can be used to micronize samples with a small amount of material, from 0.5g.
It is much more commonplace to choose a crystalline form as your candidate for development owing to preferred chemical and physical stability over amorphous counterparts as well as more trodden routes for scalable isolation and downstream processing. If sufficient bioavailability enhancement cannot be achieved using a developable crystalline form, amorphous solid dispersions (ASDs) can be a route for investigation. An ASD is a solid solution of an API within a polymer matrix. By using an amorphous phase in a solid dosage form, one can take advantage of the higher apparent solubility of the amorphous relative to that of a crystalline form to achieve high levels of supersaturation in aqueous media. An amorphous phase is less stable than a crystalline phase and therefore may crystallize. The polymer in an ASD acts as a stabiliser, kinetically trapping the amorphous phase so that it is stable over the shelf like of the finished dosage form. Some polymers may also act to stabilize the supersaturated state, allowing for the ‘spring and parachute’ effect. The spring is the supersaturated state caused by the high apparent solubility of the amorphous, the parachute is the prolonged supersaturation caused by the polymer after dissolution of the API. An ASD of piroxicam with PVP/VA (plasdone S630) was generated using our Buchi B290 lab scale spray dryer.
Dissolution is a critical step in the bioavailability of a drug, as it determines the rate and extent to which the drug is released into the bloodstream. The solubility of a drug directly affects its dissolution rate. The effectiveness of these different approaches must be compared using dissolution analysis. When developing a dissolution method, it is critical to consider what information you want to get out of it. IDR will provide a true comparison of performance irrespective of any influence of particle shape, size and morphology. Free powder dissolution (FPD) on the other hand, can provide valuable information of the impact of particle size on dissolution rate e.g., if you were to evaluate an API milled to different particle size distribution (PSD) ranges. There will be a dissolution rate intrinsic to the solid form at a constant surface area. Particle size distribution and properties will of course also have an impact on the dissolution rate. There are several experimental variables that also must be considered when developing a dissolution method including media used, agitation rate, final concentration, and concentration of surfactant, if used. We can compare the performance of different solid forms and particle engineered particles using both IDR and free powder dissolution analysis. In the present case, the initial dissolution rate of the piroxicam salt, cocrystal and the micronized API were compared using free powder dissolution using 1 % (w/v) Tween 80 and 1 % (w/v) hydroxypropyl methyl cellulose (HPMC) in simulated gastric fluid (SGF) at 37 °C. The micronized material was observed to have the highest dissolution rate under these conditions. Dissolution experiments were performed in triplicate to ensure good reproducibility between replicates. The results are summarized in Figure 3. It was shown that the micronized API had the highest dissolution rate of the different solid form and particle engineered particles assessed.
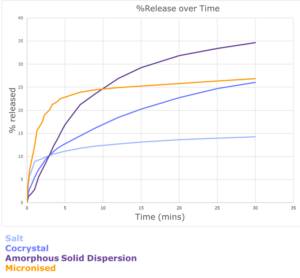
Figure 3: The initial free powder dissolution rate of micronized piroxicam, an ethanolamine salt, a succinic acid cocrystal and an amorphous solid dispersion with PVP/VA were compared. A dissolution method was developed to compare all of the candidates. It was shown that the micronized form had the best dissolution profile under these conditions.
Conclusions
Low aqueous solubility is a continuing challenge in the pharmaceutical industry. It is a priority to proactively tackle this issue during the initial stages of development. At Veranova, we facilitate this by utilizing small quantities of material to assess a number of different techniques aimed at enhancing solubility and dissolution rate. A free powder dissolution method was developed to compare the performance of a known salt, cocrystal and amorphous solid dispersion of the BCS Class II compound piroxicam with micronized material. In this case, the micronized material exhibited the best dissolution performance of the forms investigated.
References
1 Jeffrey M Ting, William W Porter 3rd, Jodi M Mecca, Frank S Bates, Theresa M Reineke Bioconjugate Chem. 2018, 29, 939−952
2 G. Steffen Paulekuhn, Jennifer B. Dressman and Christoph Saal J. Med. Chem. 2007, 50, 26, 6665–6672
About the author
Claire Wombwell is an experienced research scientist with a demonstrated history of working in the pharmaceutical industry. Since joining Veranova in August 2017, Claire has led multiple projects applying her expertise in solid state science to numerous active pharmaceutical ingredients. Her work at Veranova covers polymorph screening, salt selection, crystallization development, co-crystal screening and amorphous solid dispersion generation. Before joining Veranova, Claire worked with supercritical fluid technologies to improve the performance of medicines. Claire gained her PhD in Inorganic Chemistry from the University of Cambridge (UK).